Read the required CME information by clicking “Program Information."
Agenda:
This presentation consists of:
Patient case 1: Patient on a glucocorticoid managing their gMG symptoms
- Surveillance and management of glucocorticoid related adverse events
- Treatment of a patient with a recent MG exacerbation
- Video: Monitoring patients for steroid toxicities and adverse events
Patient case 2: Patient with gMG who is experiencing steroid toxicities
- When to implement steroid tapering
- Nonsteroidal therapies with an emphasis on the role of FcRn inhibitor
- Video: Role of the pharmacist and physician in steroid tapering and addressing knowledge gaps regarding FcRn inhibitors
Patient case 3: Starting a patient on FcRn inhibitors
- Clinical trial data of FcRn inhibitors
- Incorporating FcRn inhibitors into treatment plans
- Video: Monitoring for effectiveness and adverse events
CASE 1: MEET THE PATIENT

Brenda, a 49-year-old woman, presents to the hospital with generalized weakness and dysphagia over the past several days. She was diagnosed with myasthenia gravis (MG) 2 years ago and was found to have elevated acetylcholine receptor (AChR) antibodies. She now reports progressive weakness, diplopia, blurred vision, and dysphagia. She also describes intermittent heartburn and weight gain over the past year.
Medical history:
- Hypothyroidism
- Systemic lupus erythematosus
- Generalized anxiety disorder
- Gastroesophageal reflux disease
- Migraine
- Osteoporosis
Current medications:
- Prednisone 30 mg, oral, once daily
- Calcium/vitamin D 500 mg/5 μg, oral, once daily
- Pantoprazole 40 mg, oral, once daily
- Pyridostigmine 60 mg, oral, every 6 hours
- Sulfamethoxazole/trimethoprim 800/160 mg, oral, 3 times weekly
- Levothyroxine 50 μg, oral, once daily
Allergies:
PHYSICAL EXAMINATION FINDINGS
Vital signs:
- Blood pressure: 108/71 mm Hg
- Heart rate: 91 beats/min
- Temperature: 98.7 °F (37.1 °C) (tympanic)
- Respiratory rate: 18 breaths/min
- Oxygen saturation: 97%
- Height: 1.676 m (5 ft 6 in)
- Weight: 61 kg (134 lb)
General:
- Calm/cooperative, mild distress, acutely ill appearing and frail
Eyes:
- Normal conjunctive and sclera
Neurology:
- Best eye opening: spontaneous (4)
- Best verbal response: oriented (5)
- Best motor response: obeys commands (6)
- Glasgow Coma Scale total score: 15
- Mental status: awake, alert, and oriented to person, place, and time
- Speech: speech is nasally, somewhat weak, and slurred
- Cranial nerves: pupils are equal and equally reactive to light; some weakness when patient asked to open eyes widely. Extraocular muscle function intact, but weakness appreciated when looking left/right and down. Able to hold gaze in upward position for at least 60 seconds. No nystagmus or loss of visual fields. No facial asymmetry. Some weakness with tongue protrusion. Able to hold head up off bed and flex neck.
- Motor: follows commands and moves all extremities purposefully. Weakness worse in lower extremities than upper extremities. Worst in right lower extremity. Unable to overcome gravity in lower extremities. Able to briefly overcome gravity with bilateral upper extremities.
- Reflexes: not tested
- Sensory: intact to soft touch
- Abnormal movements: none
- Gait: deferred
Laboratory Results
- Antinuclear antibodies: negative
- Rheumatoid factor: < 15 U/mL
- Ionized calcium: 4.52 mg/dL
- Thyroid-stimulating hormone: 10.7 mIU/L
- Triiodothyronine (free): 181.82 pg/dL
- Thiopurine methyltransferase (TPMT) activity: 15.2 U/mL
- Dual-energy x-ray absorptiometry (DEXA) scan (left hip):
- Neck: -2.7
- Trochanter: -0.9
- Intertrochanter: -0.4
- Total: -0.8
CLINICAL PERSPECTIVE
Nearly 90% of patients with generalized myasthenia gravis (gMG) have comorbid conditions that can complicate their treatment.1 In one study, the most common comorbidities in patients with gMG (N = 3,516) were hypertension (42%), hyperlipidemia (37%), fatigue (25%), diabetes mellitus (18%), cerebrovascular disease (18%), hypothyroidism (15%), chronic pulmonary disease (14%), and sleep disorders (13%).2 Autoimmune disorders, including rheumatoid arthritis, systemic lupus erythematosus, and thyroid disease, also affect approximately 10% of patients with gMG.1
Unfortunately, conventional immunosuppressive therapies can exacerbate these comorbidities.2 Indeed, approximately two-thirds (67%) of patients with gMG who are treated with corticosteroids, intravenous immunoglobulin (IVIG), and/or azathioprine experience complications related to treatment, most commonly weight gain, dyslipidemia, hyperglycemia, osteoporosis-related fractures, and cataract.1
The patient’s use of prednisone (option C) is most likely contributing to her symptoms of heartburn and anxiety, as well as her weight gain and osteoporosis. Treatment-related adverse events (TRAEs) are of particular concern with long-term corticosteroid therapy, especially at high doses.3 Indeed, in a study of patients with gMG who were treated with oral corticosteroids for at least 1 year, patients experienced a mean of 2.3 TRAEs each (range, 1-6, median 2).4 The most common TRAEs were weight gain and prediabetes, each affecting 44% of patients, followed by bruising, insomnia, osteoporosis, and irritability.4
CLINICAL PERSPECTIVE
Baseline assessment and subsequent monitoring (Table 1) are critical for the safe use of systemic corticosteroids, yet many patients do not receive appropriate care.5 Among patients who received glucocorticoids for more than 1 year, fewer than 60% had their weight, glucose, or lipid levels evaluated at least once, and fewer than 25% were referred for ophthalmologic screening or a DEXA scan at least once.5
TABLE 1. Assessment and Monitoring of Adult Patients Scheduled for Systemic Corticosteroid Therapy3
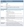
ACTH = adrenocorticotropic hormone; apo B = apolipoprotein B; BMI = body mass index; BMC = bone mineral content; BMD = bone mineral density; CBC = complete blood count; CV = cardiovascular; FPG = fasting plasma glucose; FRAX = Fracture Risk Assessment Tool; FRS = Framingham Risk Score; GC = glucocorticoid; HbA1C = glycated hemoglobin; HDL-C = high-density lipoprotein cholesterol; HPA = hypothalamic-pituitary-adrenal; LDL-C = low-density lipoprotein cholesterol; OGTT = oral glucose tolerance test; PG = plasma glucose; RA = rheumatoid arthritis; TC = total cholesterol; TG = triglycerides.
Reused with permission via CC BY license from: Liu D, Ahmet A, Ward L, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol. 2013;9(1):30.
Calcium supplements (500 mg, 2-3 times daily) and vitamin D supplements (400 IU/day) can decrease the risk of pathologic fractures during corticosteroid therapy.3 Patients should also stay current on their vaccinations, particularly for influenza and pneumococcal disease; however, those receiving immunotherapy should avoid all live or live attenuated vaccines (eg, measles-mumps-rubella, poliovirus, adenovirus, rotavirus, varicella, and nasal influenza vaccines).3,6 Yearly eye exams are also recommended, with more frequent checks for those with an increased risk of cataracts or glaucoma.3
To monitor for TRAEs during corticosteroid therapy, clinicians can use the Glucocorticoid Toxicity Index (option A), which is designed to measure change in glucocorticoid-related morbidity over time.7 The tool includes 8 domains that require a combination of laboratory tests, clinical measures, and a patient interview: BMI, blood pressure, glucose metabolism, lipid metabolism, BMD, glucocorticoid myopathy, skin toxicity, neuropsychiatric effects, and infections.7 From these, two Glucocorticoid Toxicity Index scores can be calculated: (1) the cumulative worsening score, which captures total glucocorticoid toxicity (transient and permanent) from baseline to specific follow-up points, and (2) the Aggregate Improvement Score, which measures improvement or worsening in glucocorticoid toxicity over a defined period, such as after initiating a new steroid-sparing medication.7
Several clinical tools are available to assess the signs and symptoms of MG and its effects on patients’ lives8:
- Myasthenia Gravis Activities of Daily Living scale (MG-ADL; option B), which is an 8-item measure that asks patients to report the degree of functional disability related to ocular (2 items), bulbar (3 items), respiratory (1 item), and gross motor or limb (2 items) impairment; the tool produces a total score from 0 to 24, with higher scores representing greater severity of MG symptoms
- Myasthenia Gravis Foundation of America Post-Intervention Status (option C), which combines physician global assessment with patient clinical status to measure changes in a patient’s condition after starting a particular treatment
- Myasthenia Gravis Composite score (option D), which is a 10-item tool that is used to assess the severity of MG signs and symptoms through physical examination and patient history in the following domains: ptosis (drooping eyelids), diplopia, ability to close the eyes, speech capability, chewing, swallowing, respiratory function, neck flexibility, shoulder strength, and hip mobility; each item is rated on a 4-point scale with an assigned weight, yielding a total score of 0 to 50, and higher scores indicate more severe levels of impairment
Dr. Habib and Dr. Erdman discuss the case, including managing and monitoring toxicities related to glucocorticoid therapy.
CASE CONTINUED
Brenda is admitted for monitoring and begins IVIG 1 g/kg every 24 hours (2 doses total). On hospital day 7, she starts to feel improvement in lower extremity weakness and dysphagia. Because Brenda is experiencing frequent heartburn due to the steroid, her healthcare team wants to adjust her medications.
CLINICAL PERSPECTIVE
Daily prednisone is the standard of care for patients with gMG who are experiencing worsening symptoms or myasthenic crisis.9 In this patient case, decreasing Brenda’s prednisone dose (option A) is not ideal given her recent MG exacerbation. Although the steroid will be tapered eventually, it is preferrable to start another immunosuppressive medication first.
Among options for add-on therapies for patients receiving corticosteroids, azathioprine (off-label) (option B) is an appropriate first-line immunosuppressive agent that has been shown to decrease corticosteroid dosing requirements.9 In a randomized, double-blind trial, adding azathioprine to alternate-day prednisolone reduced the maintenance dose of steroid therapy at 2 and 3 years (median value at 3 years; prednisolone + azathioprine, 0 mg on alternate days vs. prednisolone + placebo, 40 mg on alternate days; P = .02) while decreasing incidence of AEs and prolonging remission in patients with antibody-positive gMG.10 Of note, azathioprine has a boxed warning of increased risk of malignancy due to chronic immunosuppression.11 Azathioprine should not be used in patients with an absence of TPMT activity, which suggests the presence of homozygous mutations. In addition, this agent should be started at a lower dose with close monitoring during dose escalation in those with low TPMT activity(< 5.5 U/mL), which also suggests the presence of heterozygous mutations.12 Notably, low or absent TPMT activity predicts an increased risk of leukopenia in patients with gMG.12 Brenda’s laboratory results show that she has appropriate TPMT activity (15.2 U/mL), suggesting that she is a candidate for azathioprine.12
Eculizumab (option C) is not typically started on an inpatient basis and not the best choice at this time.9 Eculizumab was studied in a phase 3 randomized, double-blind, placebo-controlled study and is FDA approved for adults with gMG who are anti-AChR antibody–positive.13,14 It is contraindicated in patients who are not currently vaccinated against Neisseria meningitidis, unless the risk of delaying treatment outweighs the risk of developing infection, as complement inhibition increases the risk of infection with encapsulated bacteria such as Neisseria meningitidis.13,15 Therefore, administration of the meningococcal vaccine (both polyvalent and meningococcus serogroup B) is recommended at least 2 weeks before starting treatment with eculizumab, followed by a vaccine booster 1 month later.13 Given the presence of AChR antibodies, however, the patient may become a candidate for eculizumab in the future.
Similar to eculizumab, the complement inhibitors zilucoplan (option F) and ravulizumab (option E) are not appropriate choices at this time due to the patient being in the hospital. Patients do not typically start on complement therapies in the hospital.16 Ravulizumab is indicated for patients with gMG who are anti-AChR antibody–positive.17 Data from the phase 3 CHAMPION MG open-label extension showed improvement through week 60 in patients who received ravulizumab with least squares mean change from the randomized control period baseline in MG-activities of daily living score (-4.0; 95% CI, -4.8 to -3.1; P < .0001).18 Zilucoplan is also indicated for patients with gMG who are anti-AChR antibody–positive.19 Zilucoplan was studied in a randomized, double-blind, placebo-controlled phase 3 study, which found that patients randomly assigned to receive zilucoplan experienced a reduction in MG-ADL score compared with placebo at week 12 (least squares mean change, -2.09; 95% CI, -3.24 to -0.95; P = .0004).20 Patients should be immunized with meningococcal vaccines at least 2 weeks prior to receiving ravulizumab or zilucoplan unless the risk of delaying therapy outweighs the risk of developing meningococcal infection.17,19
Mycophenolate mofetil (option D) is incorrect because it is typically a third-line therapy for patients with gMG.9 Moreover, add-on therapy with mycophenolate mofetil has not been shown to decrease corticosteroid dosing requirements or improve disease control compared with prednisone alone.21,22
CASE CONCLUSION
Brenda begins oral azathioprine 50 mg once daily prior to discharge. She is instructed to follow up in the neurology clinic in 1 month to undergo monitoring and increase her dose as an outpatient to 2.5 mg/kg/day (150 mg/day). Her use of pantoprazole is switched to esomeprazole 40 mg once daily, and her calcium/vitamin D dose is increased to twice daily until she can be weaned from prednisone, which can occur once her azathioprine is fully titrated.
CASE 2: MEET THE PATIENT

Deborah, a 63-year-old woman with hypothyroidism, developed fluctuating leg weakness 1 year ago that resulted in difficulty standing from a seated position, climbing stairs, and walking. She developed diplopia and asymmetric eyelid ptosis 3 months later, followed by dysphagia. She also experienced worsening leg weakness that required her to use a walker and receive help with ADLs. She presented to the hospital and was empirically treated with IVIG, which resulted in symptom improvement. She did not require ventilatory support. Antibody testing was positive for AChR antibodies, confirming the diagnosis of gMG. Thyroid function studies showed treated hypothyroidism. Chest computed tomography (CT) did not show thymoma. Deborah was started on treatment with prednisone 20 mg/day and was discharged.
One month later, she exhibited significant continued oculobulbar and limb weakness at an outpatient follow-up visit and was started on treatment with azathioprine; the dose was titrated up to 2.5 mg/kg.
At today’s visit (2 months later), she mentions persistent weakness and has an MG-ADL score of 7 (range, 0-24; higher score indicates worse disease) and a Quantitative Myasthenia Gravis (QMG) score of 18 (range, 0-39; higher score indicates worse disease). She has gained 15 lb since her hospital discharge 3 months ago. Deborah expresses concern that she has not seen significant improvement in her disease course.
CLINICAL PERSPECTIVE
Identifying the appropriate next step requires an assessment of treatment options across lines of therapy, including their onset of action (Table 2).9,23
TABLE 2. MG Therapies and Onset of Action9,23

aOff-label use.
Derived from: Farmakidis C, Pasnoor M, Dimachkie MM, Barohn RJ. Treatment of myasthenia gravis. Neurol Clin. 2018;36(2):311-337; Silvestri NJ. Myasthenia gravis: making progress for more accurate diagnoses and targeted treatments. Pract Neurol. 2024;23(3):29-32.
Deborah’s weight gain—one of the most common AEs associated with steroid therapy—would rule out increasing her steroid dose and warrants a discussion about potentially reducing her prednisone dose (option A).4 Because she remains highly symptomatic, however, tapering her steroid therapy would not be appropriate at this time.
Nonsteroidal immunosuppressive agents take several months after treatment initiation to achieve benefit. For instance, the latency period for azathioprine is 6 to 18 months.9 Therefore, it would be premature to consider azathioprine failure in Deborah’s case (option B). Azathioprine should be continued, with the goal of this agent eventually becoming her primary immunosuppressive therapy. Furthermore, her prednisone dose should not be increased due to her weight gain.
Starting FcRn inhibitor therapy (option C) is a valid approach for this patient. Monoclonal antibodies that block FcRn inhibit lysosomal immunoglobulin G (IgG) recycling to reduce serum IgG, which includes levels of pathogenic IgG autoantibodies.24 FcRn inhibitors have a rapid onset of action, resulting in clinical improvement in 4 to 6 weeks, which could potentially enable prednisone dose reduction.25,26
Two FcRn inhibitors are approved by the FDA: efgartigimod and rozanolixizumab.27,28 Efgartigimod is available in both IV and subcutaneous formulations, whereas rozanolixizumab is available only subcutaneously. Both are administered via weekly dosing for a duration of either 4 weeks (efgartigimod) or 6 weeks (rozanolixizumab). Given their mechanism of action of reducing serum IgG levels by blocking lysosomal recycling of IgG, FcRn inhibitors may also reduce the levels of other IgG monoclonal therapeutics, such as rituximab and eculizumab.27,28
Another option for this patient would be to start one of the complement inhibitors (eg, eculizumab, zilucoplan, or ravulizumab). Patients starting on a complement inhibitor should be immunized with meningococcal vaccines at least 2 weeks prior to receiving this therapy.13,17,19 However, patients (such as the one in the case, who does not have a record of meningococcal vaccination) can be started on these agents while taking antibiotic prophylaxis and awaiting vaccination.16 It is important to note that there are no head-to-head trials to help decide between starting an FcRn inhibitor or a complement inhibitor, and provider preference guides which option to choose.16
Thymectomy (option D) is recommended as an early treatment option for patients with seropositive gMG who are aged 18 to 50 years to improve clinical outcomes, reduce the need for immunosuppressive therapy, and prevent hospitalization due to disease exacerbations.15 However, the benefits of thymectomy do not extend to patients older than 50 years.29 Given the patient’s age, as well as the time needed to achieve the benefits of thymectomy (≥ 3-4 months), this procedure would not be an appropriate option.9
Dr. Habib and Dr. Erdman discuss FcRn inhibitors.
CASE CONTINUED
Deborah continues both prednisone 20 mg/day and azathioprine and begins FcRn inhibitor therapy. Four weeks after completing her first cycle, her oculobulbar symptoms resolve, and her limb weakness and function improve. Her MG-ADL score has decreased to 4 (out of 24), and her QMG improves to 12 (out of 39). A second cycle of FcRn inhibitor therapy is initiated.
When she is seen for follow up 4 weeks after completing the second cycle, her MG-ADL score is now 1, and her QMG score is 5. She is no longer using a walker for ambulation and is fully independent with ADLs. She has gained 5 lb.
CLINICAL PERSPECTIVE
Oral corticosteroids are typically administered to patients with gMG using 1 of 2 approaches: (1) a high-dose, rapid-induction regimen or (2) a low-dose, slow-titration regimen.9 The low-dose, slow-titration regimen is preferred for patients with milder forms of MG, such as those with ocular MG or mild-to-moderate disease. In this approach, prednisone 10 mg/day is administered, gradually increasing by 10 mg every 5 to 7 days until a peak dose of 1.0 to 1.5 mg/kg/day is reached, up to 60 to 100 mg.9
The high-dose approach involves starting prednisone at 1.0 to 1.5 mg/kg/day, not to exceed 100 mg/day, for 2 to 4 weeks, aiming to quickly control symptoms.9 This method is particularly useful for patients with severe MG or those with significant bulbar manifestations, in whom a fast response is critical. Following the initial period, the regimen may shift to alternate-day dosing or continue with daily dosing based on the patient's condition. For milder cases, an immediate switch to alternate-day dosing may be appropriate, whereas more severe cases may require continued daily dosing. With either approach, after 4 to 8 weeks of therapy, a gradual tapering of the dose is recommended if improvement is observed.9
Although no definitive clinical guidelines exist for prednisone tapering in gMG, there is consensus that dose reduction should be accomplished slowly due to the risk of disease exacerbation (Table 3).3,15,30-32 When patients are receiving high-dose steroids (0.5-1.0 mg/kg), tapering may be performed rapidly, by up to 10 mg every 2 weeks. For patients receiving lower doses (< 40 mg), tapering should occur more slowly, by 5 mg every month. Notably, in the MGTX Study Group trial that compared thymectomy plus prednisone versus prednisone alone, patients receiving high-dose prednisone (alternate-day dose of up to 120 mg) were safely tapered by 10 mg every 2 weeks until a level of 40 mg was reached; thereafter, patients were tapered by 5 mg every month.29
For an initial steroid regimen of 15 to 20 mg/day or less, tapering should proceed more slowly, decreasing by up to 2.5 mg each month. Therefore, options A and B would not be appropriate, especially for a patient who was hospitalized a few months earlier for severe MG. Similarly, abruptly stopping prednisone (option D) could precipitate myasthenic exacerbation as well as adrenal insufficiency. Reducing the prednisone dose of 20 mg/day in 2.5-mg decrements every month (option C) is a reasonable schedule; additionally, this approach involves the administration of FcRn therapy, if needed, which has previously benefited this patient.
TABLE 3. Prednisone Tapering in Adults3,15,30-32

ACTH = adrenocorticotropic hormone; AS = adrenal suppression; GC = glucocorticoid; HPA = hypothalamic-pituitary-adrenal; ITT = insulin tolerance test.
Derived from: Liu D, Ahmet A, Ward L, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol. 2013;9(1):30; Narayanaswami P, Sanders DB, Wolfe G, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 update. Neurology. 2021;96(3):114-122; Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology. 2016;87(4):419-425; Murai H, Utsugisawa K, Motomura M, Imai T, Uzawa A, Suzuki S. The Japanese clinical guidelines 2022 for myasthenia gravis and Lambert–Eaton myasthenic syndrome. Clin Exp Neuroimmunol. 2023;14(1):19-27; Wiendl H, Abicht A, Chan A, et al. Guideline for the management of myasthenic syndromes. Ther Adv Neurol Disord. 2023;16:17562864231213240.
Dr. Habib and Dr. Erdman discuss how to taper prednisone.
CASE CONCLUSION
The patient’s prednisone dose is reduced; 6 months later, the patient is no longer on prednisone. She received 2 additional cycles of FcRn inhibitor therapy during that time.
CASE 3: MEET THE PATIENT

Roberto, a 69-year-old man with type 2 diabetes mellitus (T2DM), developed right eyelid ptosis and diplopia 3 years ago. One month later, he developed dysarthria. Evaluation led to a diagnosis of AChR antibody–positive gMG. Chest CT did not show thymoma. He started pyridostigmine 60 mg 3 times daily, experiencing partial symptom improvement. Roberto then began prednisone 10 mg/day; after 6 weeks, his symptoms resolved. However, his hyperglycemia worsened, and he required therapy escalation for T2DM.
Now, 1 year later, he has developed progressive dysarthria and dysphagia with severe difficulty swallowing food, resulting in a 40-lb weight loss. He does not have dyspnea at rest or with exertion. His MG-ADL score is 5 (out of 24), and his QMG score is 10 (out of 39). At today’s visit, the treating neurologist discusses the need to escalate therapy for Roberto’s MG.
Medical history:
- T2DM for the last 20 years, complicated by diabetic nephropathy
- Hypertension
- Congestive heart failure
CLINICAL PERSPECTIVE
Given Roberto’s history of worsening hyperglycemia after starting prednisone, a treatment approach that increases his prednisone dose (option A) is not advisable. Treatment with IVIG (option B) could be considered, but because of his history of kidney and cardiac disease, the fluid load associated with IVIG is not desirable. Either efgartigimod or rozanolixizumab are viable options, so option C is most appropriate.
CLINICAL PERSPECTIVE
Phase 3 ADAPT Trial: Efgartigimod in gMG
Efgartigimod, a modified Fc fragment derived from human IgG1, was shown to have enhanced binding affinity to FcRn receptors in preclinical models.33 The phase 3, randomized, controlled ADAPT trial compared efgartigimod with placebo in 167 adult patients with gMG.26 All patients had an MG-ADL score of at least 5 at baseline despite being on a stable dose of 1 or more therapies for gMG, and 77% had AChR antibody–positive gMG. Patients were randomly assigned to IV efgartigimod 10 mg/kg or placebo, administered as 4 infusions per 4-week cycle (1 infusion/week), repeated as needed depending on clinical response no sooner than 8 weeks after initiation of previous cycle.26
Among seropositive patients, those treated with efgartigimod had a 5-fold higher likelihood of achieving the primary endpoint of MG-ADL response (defined as a ≥ 2-point improvement sustained for ≥ 4 weeks) than those in the placebo group (Table 4).26 After the first 4-week cycle, 68% of patients in the efgartigimod group achieved an MG-ADL response, compared with 30% of those in the placebo group (OR, 4.95; 95% CI, 2.21-11.53; P < .0001). Rates of treatment-emergent AEs (TEAEs) were similar in the efgartigimod and placebo groups (77% vs 84%, respectively). The most frequently seen AEs were headache (option A) (29% vs 28%) and nasopharyngitis (12% vs 18%).26
TABLE 4. Phase 3 ADAPT Trial: Efgartigimod in gMG26

aDefined as a minimum 2-point improvement in MG-ADL score sustained for at least 4 weeks.
Reused with permission from: Howard JF Jr, Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial [published correction appears in Lancet Neurol. 2021 Aug;20(8):e5]. Lancet Neurol. 2021;20(7):526-536.
Phase 3 MycarinG Trial: Rozanolixizumab in gMG
Rozanolixizumab is a human anti-FcRn IgG4 antibody that also shows high binding affinity to FcRn receptors.33 The phase 3, randomized, placebo-controlled MycarinG trial evaluated rozanolixizumab in 200 patients with AChR antibody–positive or muscle-specific kinase (MuSK) antibody–positive gMG.25 At baseline, all patients had an MG-ADL score of at least 3 and a QMG score of at least 11. Patients were randomly assigned to undergo 6 weeks of treatment with once-weekly subcutaneous infusions of rozanolixizumab 7 mg/kg (n = 66), rozanolixizumab 10 mg/kg (n = 67), or placebo (n = 67).25
Results showed a significant clinical benefit with rozanolixizumab, as measured by change in MG-ADL score from baseline to day 43 (Table 5).25 Mean changes in MG-ADL score were -3.37 and -3.40 in the rozanolixizumab 7 mg/kg and 10 mg/kg dosing groups, respectively, compared with -0.78 in the placebo group (P < .0001 for both comparisons). The most frequent TEAEs in the rozanolixizumab 7 mg/kg and 10 mg/kg groups, respectively, were headache (45% and 38%), diarrhea (25% and 16%), and pyrexia (13% and 20%).25
TABLE 5. Phase 3 MycarinG Trial: Rozanolixizumab in gMG25

LS = least-squares.
Derived from: Bril V, Druzdz A, Grosskreutz J, et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive phase 3 study [published correction appears in Lancet Neurol. 2023 Oct;22(10):e11]. Lancet Neurol. 2023;22(5):383-394.
Other Investigational FcRn Inhibitors
Additional FcRn-blocking agents undergoing evaluation in ongoing phase 3 trials of patients with gMG include nipocalimab (NCT04951622) and batoclimab (NCT05403541).33
Dr. Habib and Dr. Erdman discuss FcRn inhibitor treatment and monitoring patients.
CASE CONCLUSION
Roberto is started on an FcRn inhibitor and experiences improvement of his dysphagia.


 Med-IQ is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education. 1.0 contact hour (0.10 CEUs) of credit for pharmacists/pharmacy technicians. ACPE #0476-0000-24-006-H01-P. This knowledge-based activity is designed for all pharmacists.
Med-IQ is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education. 1.0 contact hour (0.10 CEUs) of credit for pharmacists/pharmacy technicians. ACPE #0476-0000-24-006-H01-P. This knowledge-based activity is designed for all pharmacists.