ReachMD
Be part of the knowledge.™Targeting Defective Tau Proteins May Be Needed to Treat Alzheimer’s Patients
 news-medical.net
news-medical.net
In November, researchers reported the drug lecanemab slowed the progression of Alzheimer's disease. The effect was modest, but it has generated tremendous excitement because it was the first time a drug had been shown to be able to affect the course of this relentless, incurable disease.
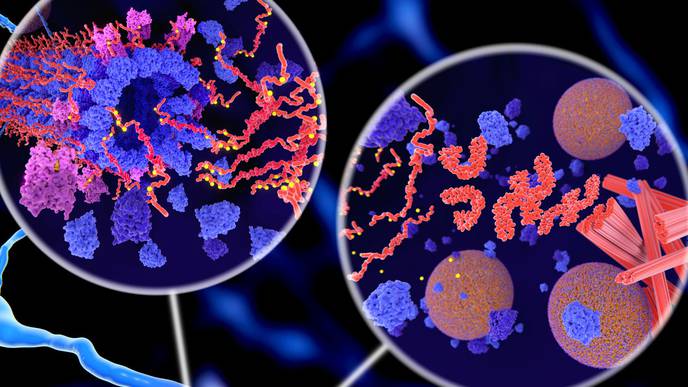
The drug, lecanemab, is a manufactured antibody that helps clear an abnormal protein called beta-amyloid, which forms insoluble clumps called amyloid plaques around brain cells. It is thought that amyloid initiates and sustains the destruction of brain cells that leads to the cognitive decline and eventual dementia that afflicts Alzheimer's patients.
But many researchers believe that for any treatment to have a major impact on the course of Alzheimer's, they will also have to target a second protein that to date has not received as much attention as beta-amyloid, a protein called tau.
The amyloid plaques start the disease cascade, so it makes sense to try to eliminate them, but it's tau that kills the cells."
Brian Kraemer, Professor of Medicine, Division of Gerontology & Geriatric Medicine, University of Washington School of Medicine
Kraemer specializes in neurodegenerative diseases caused by tau, called tauopathies. These include a long list of incurable neurodegenerative diseases. In some, abnormal tau appears to be the primary cause of the disorder. These are called pure tauopathies. They include frontotemporal lobar degeneration, progressive supranuclear palsy, and Pick's disease. On the other hand Alzheimer's is called a mixed tauopathy, because beta-amyloid plays a role.
Tau, which rhymes with "wow," stabilizes crucial structures within cells, called microtubules. These structures serve as a cell's internal skeleton and act as conduits through which the cell shuttles material from place to place.
In Alzheimer's disease and other tauopathies, tau is defective. It detaches from the microtubules and forms insoluble aggregates within cells called neurofibrillary tangles. Breakdown of the microtubules and accumulation of neurofibrillary tangles disrupt the brain cell's ability to function and eventually leads to cell death.
"If we were to target any one thing in Alzheimer's disease, we probably should be targeting tau," Kraemer said. "It's the most closely tied to the decline in cognitive dysfunction. You want to get rid of amyloid but what you really want is preservation of cognition. That requires targeting tau."
In an article published in the journal Proceedings of the National Academy of Sciences Dec.26, Kraemer, his team, and lead author Randall Eck, a student in the UW Graduate Program in Neuroscience, report the identification of a protein that appears to be crucial in forming abnormal collections of tau. The scientists showed that by blocking the gene required for the production of the protein it is possible to prevent the accumulation of tau in an animal model.
The protein is called speckle-type POZ protein (SPOP ). The name refers to how it is found in speckle-like compartments in the cell and the fact that it contains a particular stretch of amino acids known as a POZ domain. This is one of several proteins Kraemer and his coworkers have linked to tauopathies. Another protein, called SUT-2 for suppressor of tauopathy-2, is being explored for its therapeutic potential.
The exact role the protein plays in diseases involving tau is not clear. But it appears to be involved in an essential process by which cells handle and eliminate defective proteins. The findings suggest that if drugs could be developed that inhibit the effect of this protein it might be possible to treat Alzheimer's disease and other tauopathies.
To identify these key regulatory proteins, Kraemer and his coworkers use an animal model his lab created two decades ago. The model is a genetically engineered version of a small worm, normally found in the soil, called Caenorhabditis elegans, or C. elegans for short. C. elegans lives for only about three weeks, so it is ideal for studying how gene mutations affect an organism's growth, development and function throughout its lifespan.
To create the model, Kraemer and his team introduced the human gene for the tau protein into the roundworms.
In their experiments, the scientists have demonstrated that the altered worms develop many of the abnormalities seen in human tauopathies: the accumulation of insoluble tau, progressive nerve cell death, behavioral deficits and shortened lifespan.
The researchers then conducted a screen of all the genes in the worm to see if randomly knocking out any of them could prevent these changes. This approach led them to first identify the gene for SUT-2 and more recently SPOP.
"When we eliminate the SPOP protein in our tau worm model, we see a dramatic decrease in the accumulation of tau and progressive nerve cell death as well as an improvement in behavioral deficits and lifespan," Eck said.
Kraemer, Eck, and other researchers in the field are now looking into whether their findings in this C. elegans model can be translated into treatments in humans. The first step is to see if suppressing these genes can have a similar protective effect in a mouse model of the disease. Studies suppressing the gene for SUT-2 are promising and studies looking at SPOP are underway.
"We're still very much in the early days of developing effective disease-modifying drugs for Alzheimer's," said Kraemer. "A tau inhibitor may be enough to treat pure tauopathies, but for Alzheimer's I think we're going to have to hit both tau and amyloid to have an effective treatment."
Facebook Comments